


The Curious Case of Ebola's Phylogenetics, Part II
See the forest through the phylogenetic trees

Modern science is path dependent. What new papers say depends on what the papers that preceded them say. The ability to get new papers published depends on what related papers were able to get published before them, and who their authors were. Modern science is a more catty subfield of fashion, and with a much smaller market cap.

The first part of this series demonstrated that the phylogenetic literature resulting from the 2014 Ebola outbreak in West Africa does not methodologically resemble other emerging disease literatures. A review of 80 or so papers shows the phylogenetic methods used in the Ebola literature are markedly different from the others, and moreover, they seem to have been disproportionately influenced by the early authors who first published in 2014.
This installment of the series will be broken down into four sections, one for each of the four earliest papers in the Ebola literature, in order. We will scrutinize each, some in more detail than others, and where appropriate we will reproduce some of the phylogenetic results and examine whether the primary methodological choices in these papers are justified.
Baize et al., 2014

By comparison to other papers in this review, this is an interdisciplinary paper—part phylogenetic analysis, part contact-tracing, clinical investigation. The latter component is probably the reason it landed in The New England Journal of Medicine. This is the first paper to examine the three very first Ebola virus cases confirmed by genetic sequencing in middle to late March of 2014. The accessions are here, here, and here. It is these sequences which will form the data backbone of the rest of this poast.
The paper uses contact tracing and medical records to putatively track suspected cases back through confirmed cases and concludes an infant who died in a Guinean village in December 2013 was the index case. In a SARS-COV-2 world this methodology isn’t as crazy as it sounds since Zaire Ebola is spread by direct contact with the bodily fluids of an infected person. And yes, you did read that right: there were not test-kit confirmed cases for almost three months after the identified index case.
Phylogenetically, the authors reach a surprising conclusion:
Phylogenetic analysis of the full-length sequences established a separate clade for the Guinean EBOV strain in a sister relationship with other known EBOV strains. This suggests that the EBOV strain from Guinea has evolved in parallel with the strains from the Democratic Republic of Congo and Gabon from a recent ancestor and has not been introduced from the latter countries into Guinea.
So the Makona strain that emerged in West Africa is divergent from known EBOV strains. It’s a sister strain. The authors produce this phylogenetic tree in the manuscript, rooted by distant ebolavirus outgroups.
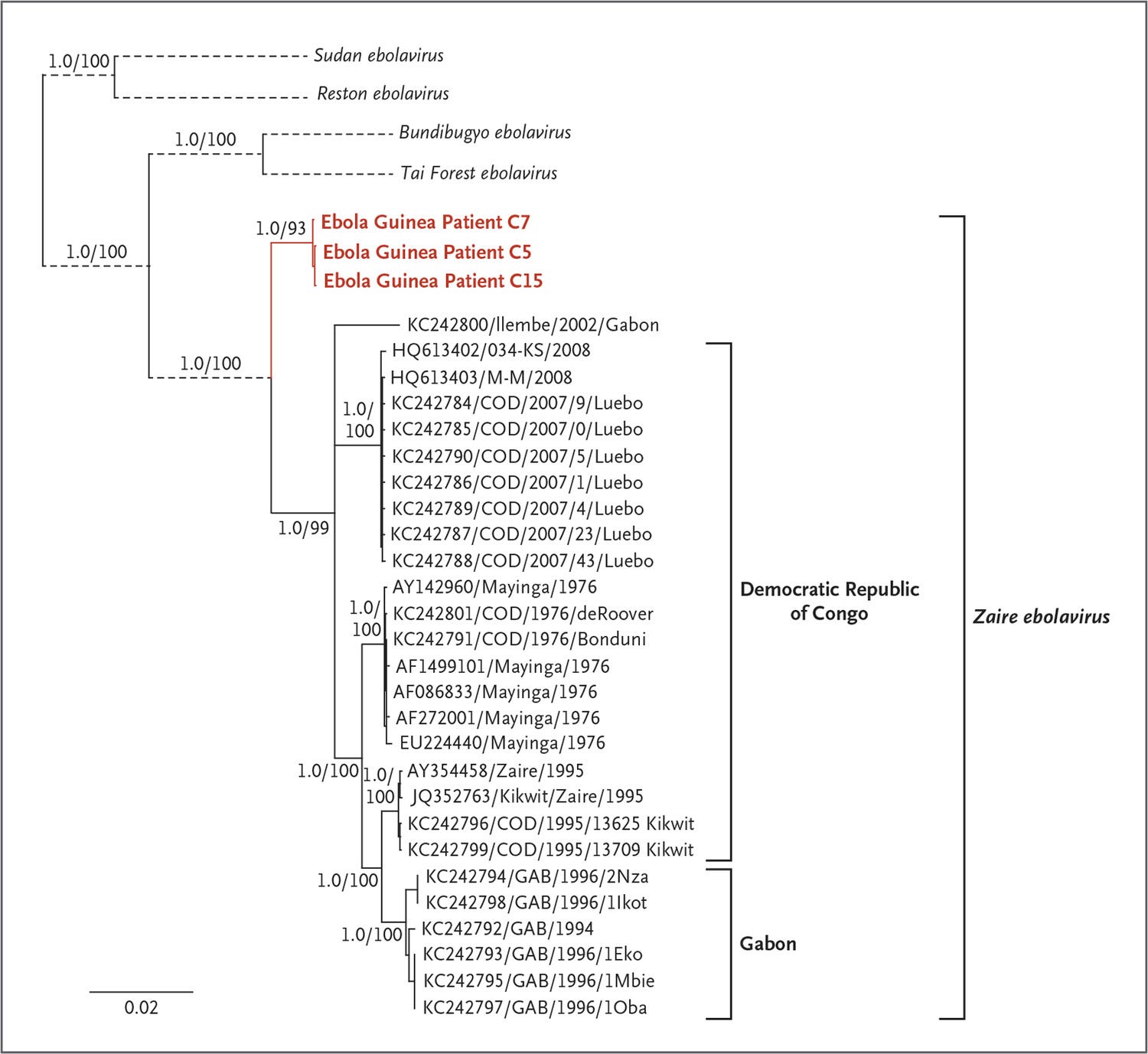
This is where I am a little over my skis. From what I can tell, the scientific consensus that exists to this day is that all human Zaire ebolavirus infections result from a single spillover that occurred in Zaire in the 1970’s and which has played peek-a-boo with clinicians and virologists ever since.
The disease emerged. It was extinguished. It emerged again, as a daughter of the virus before it, and was dealt with again. Only daughters of the initial virus cause human infections. Cousin viruses do not. There have been no other independent introductions, across all of Africa.
This is a stunning thesis to defend. It contains multitudes.
(Informed persons: if I have misstated the case here, please correct me. It seems unbelievable.)
In Fig 3, Baize et al. apparently challenge this long held notion. To their credit, in their supplementary material they allow for the fact that they may be mistaken.
They plot two possible trees. Their respective tree shapes depends on the evolutionary rate of ebolaviruses.

Under fast molecular evolutionary scenarios, the novel Makona strain in West Africa looks pretty normal. Far up the tree, but not super far. Under slower evolution, the Makona strain is a basal, sister lineage, new and unexpected.
Dudas and Rambaut, 2014

For such a technically dense outcome, this is a surprisingly short paper. Dudas and Rambaut is a critique of the Baize et al. finding that the novel Makona strain from West Africa is a sister lineage to known EBOV strains.
We report evidence that points to the same Zaire ebolavirus lineage that has previously caused outbreaks in the Democratic Republic of Congo, the Republic of Congo and Gabon as the culprit behind the outbreak in Guinea.
They assembled a collection of EBOV and outgroup sequences and created an alignment for their own phylogenetic analysis. The .nex files are checked in here but astonishingly, the mrbayes code is not. I forked the repository here. This is the dataset I’ll be using for the rest of this analysis.
Dudas and Rambaut reproduce the Baize et al. finding that Makona is a EBOV basal sequence on the full genome alignment. I also am able to reproduce this to an appropriate degree of accuracy when forcing the EBOV clade as the ingroup in Figure 6. .nex file with mrbayes code here and tree here.


Dudas and Rambaut reject this phylogeny. The crux of their argument seems to be that inconsistent sampling and the distant phylogenetic outgroups chosen by Baize et al. like Restonvirus and Sudan ebolavirus have created a situation where the Makona strain is artificially drawn deeper into the tree by standard phylogenetic methods. Although they don’t use it, the term for this phenomenon is “long branch attraction” or LBA.
As far as I can tell, reference to this statistical problem in phylogenetic analysis is unprecedented in viral literature. It doesn’t appear anywhere else in the review I covered in Part I. It may even be unprecedented in all emerging disease literature. I couldn’t find a reference to it anywhere in a research paper apart from Calvignac-Spencer et al., which we’ll examine later. I had to ask an ecologist friend of mine whether any practicing investigator has ever even heard of this. They said, yes, this is a known issue, and pointed me to an incredibly detailed twenty year old technical review of this issue, Bergsten, 2005. This review is also subsequently cited by Calvignac-Spencer et al., and more importantly, cited by wikipedia.
Bergsten apparently knows absolutely everything there is to know about LBA. I emailed him to ask whether the situation described in Dudas and Rambaut is an example of LBA, but he did not reply to me. In the absence of word from the Sage Himself, we have to use his review to scrutinize this issue more closely.
Bergsten has an entire section devoted to methods of detecting LBA. We’re going to use three of them to study whether or not the phylogeny Dudas and Rambaut reject actually is an example of this phenomenon.
Separate partition analysis
On this method Bergsten states
… comparing the results from different genes evolving at different rates has also been applied in order to see if a fast gene might show signs of grouping long branches together, in contrast to slower evolving genes (Moreira et al., 2002). Although suffering from the same problem as those above, i.e., it does not consider that gene trees do not need to match species trees and thus not each other, it could be indicative given that the problem of LBA should preferentially occur at higher rates of evolution (Huelsenbeck and Lander, 2003).
Interestingly, Dudas and Rambaut don’t actually cite Bergsten, but they use this method to test the tree topologies. They break the whole genomes into separate partitions: intergenic regions and coding regions, and rerun the analysis separately on each one. Due to the evolutionarily conserved nature of coding regions, the intergenic regions evolve faster. This partitioning of the genome will provide a good test for LBA since the tree topologies can be compared across the genomic partition. If LBA exists in this dataset, Bergsten thinks the Makona rooting will be deeper for the faster-evolving intergenic region and shallower for the more conserved protein-coding regions. OK, so what happens?
I was able to reproduce this part of the paper as well. The intergenic phylogeny.


And the protein-coding phylogeny.


Wait, BOTH partitions place Makona in the EBOV clade? That’s not what Bergsten said would happen. He said if there was long branch attraction, only the protein coding analysis might make Makona fall in the EBOV clade and the intergenic tree would maintain a deeper place for the new Makona strain. Dudas, Rambaut, and I agree this isn’t actually happening here. In other words, the authors’ own test doesn’t actually demonstrate what they claim, which is that the long-branched Makona strain is incorrectly being drawn to the long-branched outgroups.
I’m not completely sure what’s going on here, but it strains my belief that a reviewer for this paper would not notice it. Yes, under the partitioned strategy, both analyses show Makona within the EBOV clade, but that’s not actually what’s supposed to happen if LBA is present. Not the flex they think it is.
Long-branch extraction
So separate partition analysis was a bust. Let’s try Bergsten’s favorite LBA test, long-branch extraction. He states
Siddall and Whiting (1999) and Pol and Siddall (2001) suggested a simple test for cases where LBA is suspected to be a problem. They noted the obvious fact that for a long branch to be able to attract or be attracted there needs to be another long branch simultaneously in the analysis. So, in a case where two long branched taxa are grouped together, removing one while keeping the other in and vice versa would allow them to find their correct position in two separate analyses. If the clade was correct, then the separate analyses would not alter the position of the long branches in the tree. If however, one branch “flies away” to another part of the tree then it would be suspected that the clade was an LBA artifact.
Dudas and Rambaut never described doing this test, so I can’t cite their analysis, but I conduct my own.
This is a little tricky to do. As you can see, there are multiple divergent clades which might serve as outgroups and one or more of them could be attracting the Makona strain and messing up the analysis, so it’s certainly not just two separate analyses that need to be done.
Remember, Dudas and Rambaut conclude
We believe that at present no suitable outgroup sequences to root the EBOV phylogeny exist and that a temporal rooting gives the most consistent results.
So not only is a single outgroup creating LBA, but apparently none of them are appropriate. This is a very strongly worded claim, and it’s after
This shows that the rooting of this clade using the highly divergent other ebolavirus species is very problematic.
Lots of words ending in “y”.
Using Bergsten’s long branch extraction method, is there any merit to these claims?



Figure 9 is the phylogeny reconstructed with the RESTV, SUDV, and TAVF clades excluded, respectively. Only with the SUDV exclusion analysis does the Makona strain become a sister strain to EBOV. With the other exclusion analyses, it stays nested in the EBOV clade. To be extra careful, I also did this where these long-branched outgroups were excluded in pairs, RESTV/SUDV, RESTV/TAVF, and SUDV/TAVF.



Again, the evidence looks mixed. In two out of three of these alternative outgroup exclusions, Makona isn’t drawn out to be a sister EBOV clade. Only when SUDV and TAVF are excluded does Makona become deeper in the tree.
Another thing to note is in Figure 11. LBA makes long branches attracted to each other, so outgroups can be attracted to long ingroup branches as well. I created a phylogeny excluding the Guinean Makona cases, to see if the outgroup topology changed with it.

Clearly, the outgroup topology hasn’t changed, so Makona isn’t obviously attracting outgroups inward.
Overall, I’d say the evidence is mixed here. You could perhaps provisionally conclude that RESTV and only RESTV is a long branch attracting Makona deeper up the phylogeny, but I’m not sure. Figure 11 shows no inverse effect of Makona pulling the outgroups in. There’s no way one can conclude that
We believe that at present no suitable outgroup sequences to root the EBOV phylogeny exist
Artificial LB sequences
The final test I’ll do on this is the inclusion of a randomly generated sequence into the phylogeny. Bergsten states
Several studies have tested LBA by creating artificial taxa with random (long-branched) sequences (Sullivan and Swofford, 1997; Philippe and Forterre, 1999; Stiller and Hall, 1999; Qiu et al., 2001; Stiller et al., 2001; Graham et al., 2002), and this approach dates back to Wheeler (1990), who showed that a random sequence is expected to attach to a phylogeny on the longest branch. Wheeler's concern was that the use of a too-distant outgroup will act as a random sequence and artificially root the ingroup on the longest branch.
… The test of LBA functions as follows; create a number of (e.g., 100) random sequences of the same length as the original. Exchange the real outgroup with the artificial sequences one by one. Run a parsimony analysis for each exchange and compare them where the tree is rooted with the root of the original analysis using the real outgroup sequence. If the tree is rooted at the same place with the original outgroup as with a high percentage of the random sequences, then it is suspected that the rooting is not based on a phylogenetic signal but rather on LBA artifacts.
So if the topology of the tree does not frequently change from the outgroup induced topology when we include synthetic sequences, we can conclude that LBA is a least a plausible issue in the dataset. I created ten artificial sequences from nucleotide frequencies observed in Dudas and Rambaut’s dataset (C: 0.218, G: 0.196, A: 0.316, U: 0.269, and R: 2.725e-6), excluded all the other real potential outgroups, and created ten new EBOV phylogenies with the ten synthetic sequences.
I actually think this is a pretty weak, second or third best test, and Bergsten agrees, but in 9 out of 10 of the randomly created sequence analyses, the Makona strain was placed next to EBOV on the phylogenetic tree, which is some evidence for LBA.
Closing remarks
The case that Dudas and Rambaut make themselves in this paper is weak. The separate partition analysis they perform is not consistent with a long branch artifact present in the phylogeny. It only shows a more shallow Makona strain if you slice the data a slightly different way than Baize et al. do it. They don’t do a better analysis. They might actually be correct that LBA is contributing to the deep phylogenetic placement of Makona in Baize et al., but the paper hastily concludes that; it doesn’t demonstrate it.
Unfortunately, the tests Bergsten recommends seem highly inconclusive to me in this case.
As a separate issue, I just don’t think some of things they say in this paper make any sense. The paper concludes that the likeliest date of this Makona strain into West Africa is 2002, but they also state
This approach indicates that the outbreak in Guinea is likely caused by a Zaire ebolavirus lineage that has spread from Central Africa into Guinea and West Africa in recent decades, and does not represent the emergence of a divergent and endemic virus.
Why is a virus that’s been in the jungle in West Africa a dozen years not “endemic”? Why is a virus that confuses the phylogenetic analysis after living in West Africa for a dozen years not “divergent”? Unanswered questions.
Early in the paper they state
The branch leading to the Guinea outbreak is long, not because it is a divergent lineage but because it is the most recently sampled so has had the most time to evolve.
In fact, this was true of every single ebolavirus outbreak before the West African one in 2014. The most recently sampled virus is by definition the observed one that’s had the most time to evolve. Why does this mean this strain is not divergent? Unclear.
Calvignac-Spencer et al., 2014

This paper concurs with Dudas and Rambaut’s conclusions that the novel strain of Ebola in West Africa was actually a member of the Zaire ebolavirus clade, and make explicit the allegation that LBA is the cause.
Dudas and Rambaut (2014) recently demonstrated that improper rooting can end up in supporting strikingly erroneous evolutionary scenarios, which can in turn mislead the formulation of important epidemiological hypotheses2 .
…
Accordingly, Guinea 2014 EBOV is more likely to be the result of a fairly recent introduction of ZEBOV from Central Africa than a long-term endemic in West Africa.
The initial misplacement of Guinea 2014 EBOV was due to unnoticed long-branch attraction to the outgroup4 . As pointed out by Dudas and Rambaut (2014), this phenomenon made the long branch of Guinea 2014 EBOV drift towards the basis of the Zaïre clade as it was attracted to the other (very divergent) EBOV lineages included in the analyses (Bundibugyo, Taï Forest, Reston and Sudan)2 .
They applaud the use of a temporally-driven rooting for the EBOV clade, despite the fact this had never appeared in the ebolavirus literature before. The paper does cite Bersten’s review, but they don’t describe doing any of the LBA tests I highlighted above or even add any deeper LBA analysis on top of Dudas and Rambaut’s . The other papers Calvignac-Spencer et al. cite (Carroll et al., 2013 and Lauber et al., 2012) do not do the long branch analysis on pre-Makona strains. In the absence of strong evidence for long branch artifacts (which I’d again like to point out had never really been considered in viral phylogenetic literature, and certainly not in ebolavirus literature), why exactly is the novel Makona strain being a deep branch a “strikingly erroneous evolutionary scenario”?
Little enough is known about ebolavirus. Even less is known about how ebolavirus got to West Africa in the first place. You use data like this to discover what you don’t know. You shouldn’t categorically rule out data driven scenarios, or be incautious about stating what’s known.
Gire et al., 2014
I’ve actually decided a quick review of this paper is more appropriate for Part III. Stay tuned. It will be an event.
This is a very interesting analysis and seems to show attempts to nudge the trees to a preferred outcome. This reminds me of the attempts by Rambaut et al, to push away RaTG13 as the closest related taxon to SARS2, instead claiming that the BANALs are closer (which happily come from Laos as opposed to S.China in the case of RaTG13). Standard phylogenomic trees show RaTG13 as the sister species to SARS2, confirmed independently by several groups. Rambaut et al had to invent the synthetic recCA outgroup in order to move RaTG13 away from SARS2 on a phylogenetic tree
'Under slower evolution, the Makona strain is a basal, sister lineage, new and unexpected.'
Rambaut took efforts to challenge the slow evolution viewpoint, probably to support the claim that the Makona strain was not a divergent lineage, in the following 2016 Comment:
https://www.science.org/doi/10.1126/science.aaf3823