


The Curious Case of Ebola's Phylogenetics, Part I
After the 2014 outbreak in West Africa, a changed scientific literature emerged

Until 2014, Ebola virus was unknown to West Africa. In a part of the world known for rickety health infrastructure, authorities were caught unprepared for the unprecedented and uncontrolled spread of a disease with which they felt they might never have to contend.
As with atypical outbreaks before and after this one, the Ebola outbreak, reported to have started in Guinea, was quickly analyzed by western scientists and eventually contained by dogged local effort, western containment known-how, and the deaths of many volunteer healthcare workers.
This is not a story about hardship and sacrifice. Doctors Without Borders has already covered that. It’s technical analysis of the scientific papers that emerged during and after the crisis. The scientists who originally studied the West Africa Ebola outbreak spawned a literature suspiciously unlike those before or after it. I’ll show you how.
Part I of this series of three will be a systematic review of phylogenetic methods across four different emerging disease literatures: MERS in 2012, Ebola in 2014, Zika in 2015, and Moneypox in 2022. In all these examples, a viral disease was identified in a geography which was unexpected given then historical circumstances and was analyzed with the power of modern mass sequencing technology. Scientists used this sequencing to infer the phylogenetic place of the novel outbreak. In this review, we will see how the papers in the wake of the Ebola 2014 are different from the other three emerging viral disease literatures.
Part II will be a deep dive into why the Ebola 2014 phylogenetic literature is different, and why that difference is not justified. The early authors Dudas and Rambaut, Calvignac-Spencer et al., and the summer blockbuster Gire et al. seem to have technically steered the literature in a way that makes the Makona strain of Ebola that overran West Africa look more like its older Ebola relatives and less like an unrelated phenomenon in need of a novel explanation. You will learn about long branch attraction and why it’s important. This will be wonkish, but unfortunately, it’s the only way to build the argument leading up to Part III.
Part III will be more speculative. I’ll tell you what I actually think of all this. I’ll discuss the potentially problematic motives of the scientists involved in this literature, and give some potential explanations for why the Ebola literature turned out differently than it might have. I also want to leave some room for uncertainty. My thesis might be incorrect and I’m hoping to garner some interesting discussion.
Background
Everyone has seen a phylogenetic tree. Even before there was sequencing or DNA or Nespresso, famous people drew them.
Darwin would have used guesses and hunches, geography and morphology to do this, and there was nothing truly “genetic” about his phylogenetic tree. There was no molecular evidence undergirding it.
Phylogenetics is one of the most important tools in modern virology for several reasons.
Sequencing is now absurdly cheap, and can be done at massive scale.
The nature of viral outbreaks can guarantee enormous amounts of full-genome sequencing data, and viral genomes are small compared to many others.
Modern compute constraints guarantee these full genomes can be readily analyzed.
There’s not an inexpensive way to assess “viral morphology”, if that’s even a thing. Rigorously studying viral phenotypes requires detailed scientific experimentation.
There are two types of phylogenetic trees: rooted and unrooted. Darwin’s was unrooted: he suspected that all life emerged from a single source, but he didn’t have much of a clue about what that root actually was. Unrooted trees can describe relationships between genetic sequences, but there’s no time information about the how sequences in the tree evolved. Evolution obviously occurs over time, which goes in a single direction, so if you want to study evolution of a species over time you need a way to root the tree.
The rooting sequence is an educated guess about which sequence is most ancestral to the others in a phylogenetic dataset. There are different ways to do the tree-rooting, and there are many different ways to fit a phylogenetic tree to dataset of genetic sequences. An investigator makes a decision about which rooting method and which phylogenetic algorithm will work the best in certain circumstances.
Rooting
There are several established ways to root a phylogenetic tree.
The “outgroup” method. If there’s a new phylogeny to study, an investigator will pick an outgroup sequences or sequences that are suspected to be related to the new phylogeny, but are separated either by time, by geography, or by sampling circumstances from the new sequences. The idea is that the outgroup will have evolved in some degree of isolation from the new viral sequences, and is therefore an appropriate proxy for an ancestor of the new phylogeny. Alien phylogeneticists, with limited knowledge of the history of human evolution, would choose chimpanzees or gorillas as an outgroup in studying human phylogenetics. Lyons-Weiler et al. is the definitive technical account about how this works.
The “temporal" method. This rooting method exploits the fact that viral sampling is often timestamped with a high degree of fidelity. Even in fast-moving viral outbreaks, a timeline of sequences can be established, and if an investigator trusts the sampling procedures, a rooting sequence should logically just be the earliest one. This logic can be extended even to viral samples that were gathered decades ago. As justification for this method, investigators will often make sure the genetic divergence of the samples corresponds to the sampling dates (a so-called root-to-tips regression). Newly arrived alien phylogeneticists, in their study of human evolution, would likely not be able to use this method because they would have insufficient sampling fidelity back through time.
The “midpoint” method. This is less commonly used, but does occur in the viral phylogenetic literatures on occasion and is worth mentioning as an alternative to the above methods. The investigators basically infer “the middle” sequence of the phylogeny, assume that viral evolution proceeds similarly on all the branches of the tree, and set that middle sequence as the root. Viral evolution can undergo severe bottlenecks and challenges to the assumptions of this method. Alien phylogeneticists would probably not use it.
Systematic Review
I conducted a systematic review of the rooting methods used in the phylogenetic analyses in four separate emerging disease literatures. This consisted of 82 different papers across 4 different viral outbreak literatures: MERS in 2012, Ebola in 2014, Zika in 2015, and Moneypox in 2022. In all these papers, I determined which rooting method, if any, was used to determine where the new virus existed in established phylogenies. A Google Sheets version of this review exists here, and the review is also checked into github here.
The criteria I used to select these four literatures were as follows:
Large scale viral sampling and sequencing had to be technically feasible at the time of the outbreak. This wasn’t really the case with SARS1 or previous Ebola outbreaks.
Each outbreak had to be geographically unusual or exhibit novel epidemiology. MERS emerged in the Middle East, distantly removed from bat colonies in Southeast Asia. In 2014, Ebola emerged in West Africa far away from the Congo Valley which was its historical domain. Before 2015, Zika had never been observed in the New World. In 2022, Monkeypox had been endemic to parts of West Africa but exhibited a novel pattern of spread in western countries.
Each literature needed to be sufficiently detailed and mature.
Each literature needed to not be SARS2 because my personal life and sanity are important.
If you think there are other outbreaks that should be included in this analysis for comparison please let me know. No, I’m not including SARS2, not matter what you say.
The summary statistics for the systematic review looks like this. To the authors whose methods sections were too incomplete for me to deduce what rooting type was being used, forcing me to code it as “unclear”, I hope you feel a little badly about yourselves.
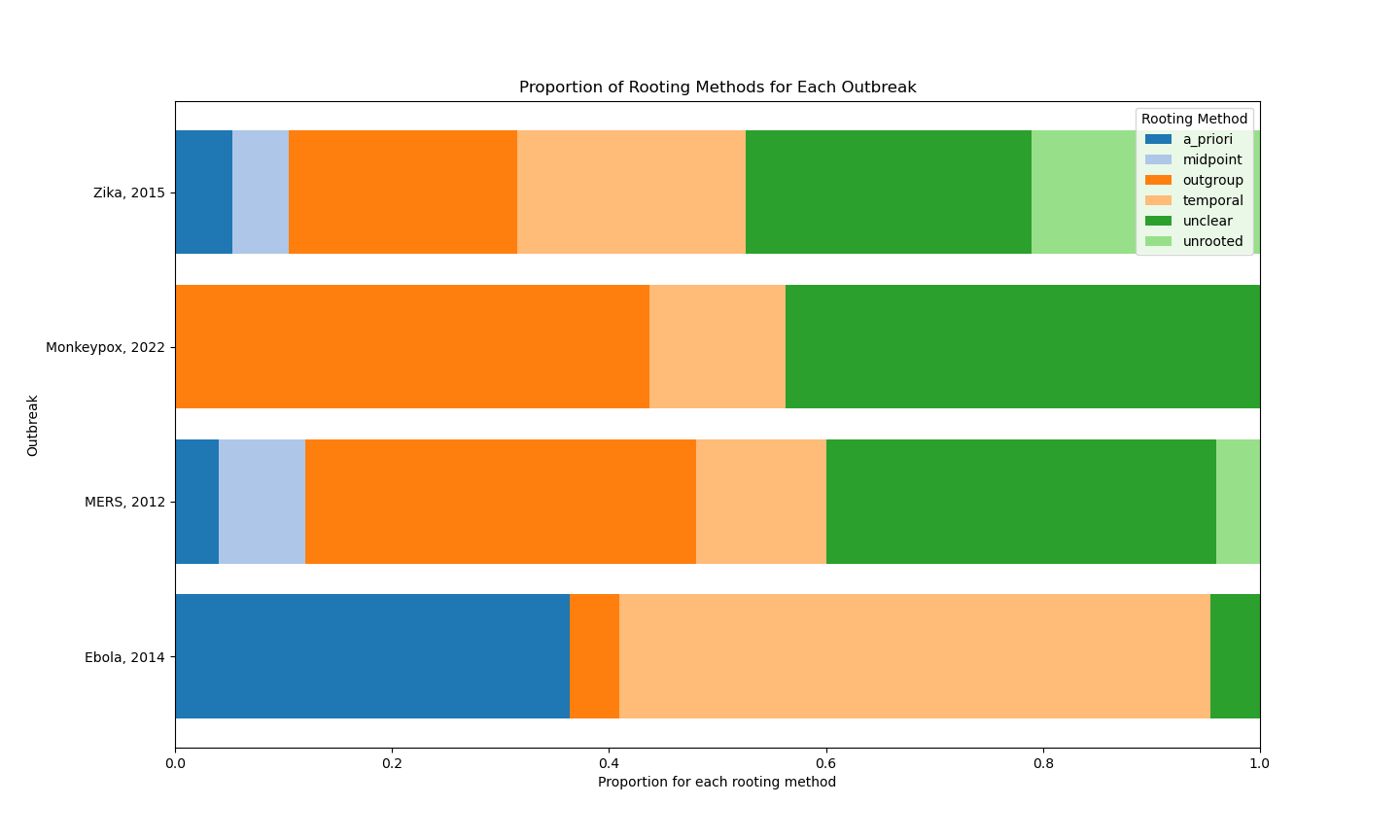
The methods dominating the phylogenetic literature that resulted from the 2014 Ebola outbreak in West Africa are very different from the methods from other emerging viral disease literatures.
Outgroup rooting is spurned for temporal methods. There’s much less methodological diversity. There’s much more reliance on previous papers. “a_priori” is the code I used for assuming previous analyses are correct a scholarly citation of a previous paper.
Before 2014, modern Ebola virus papers (Walsh et al., Wittman et al., and Grard et al.) use outgroup rooting. Baize et al. is the first published paper in this Ebola literature and is the only one to use outgroup rooting after and during the 2014 outbreak. We’re going to find out much more about why that’s the case in Part II.
In addition to the developed state of each literature observed in Figure 2, it’s easy to see that as time passed the Ebola 2014 literature unfolded very differently from the others. Here is the Ebola literature sorted by publication date.
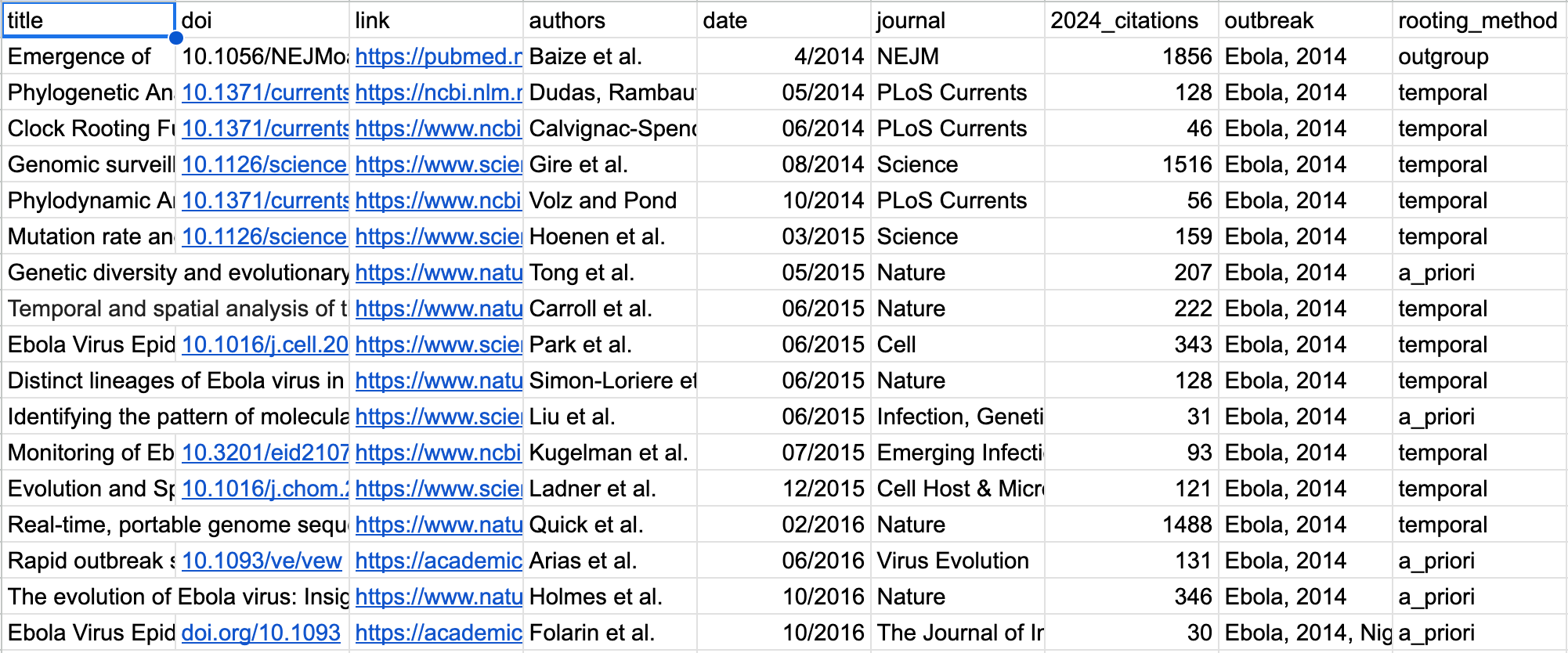
Compare this to the Zika literature starting in 2015.

And to Monkeypox in 2022.

Very quickly there developed a consensus around temporal rooting as the preferred methodology for creating the phylogenetic trees in Ebola 2014 that did not occur in other emerging viral disease literatures.
Latham and Husseini are also perplexed by this, though perhaps not in the quite the same terms I am.
… even though many of these papers were published in the foremost scientific journals, like Science (Gire et al., 2014; Hoenen et al., 2015), Nature (Carroll et al., 2015; Simon-Loriére et al., 2015; Quick et al., 2016; Tong et al., 2016; Holmes et al., 2016; Dudas et al., 2017) and Cell (Park et al., 2015), their conclusions, that the epidemic began in Guinea are unsound due to this circularity and failure to make use of an out-group.
The circularity of relying on timing alone was surely known to the senior authors (and presumably to peer-reviewers also). … Why then was the obvious step of testing the Guinea root with an out-group skipped in all these papers? …
We are not in a position to remedy that omission. However, what we can say is that not corroborating the clearly flawed clock method with the obvious test is a very puzzling and troubling omission…
I am hoping to remedy this omission. In Part II, there will be .nex files and mrbayes. We’ll find that the authors in the early 2014 Ebola literature, particularly Dudas and Rambaut and Calvignac-Spencer et al., disqualify outgroup rooting by appeal to a technical argument around a phylogenetic phenomenon called “long branch attraction” . No, this is not a tree fetish. We’ll explore how justified this technical argument is.
I’m pretty sure I don’t understand this, but nonetheless this made me laugh:
“To the authors whose methods sections were too incomplete for me to deduce what rooting type was being used, forcing me to code it as “unclear”, I hope you feel a little badly about yourselves.”
On to Part 2…